
Introduction
GDP audits carry consequences that can end a pharmaceutical distributor's business overnight. License suspensions, costly product recalls, and import/export bans are not theoretical risks — they're enforcement actions that regulatory bodies issue regularly. A 2025 FDA Warning Letter to Sterling Distributors cited failures to investigate suspect products and verify authorized trading partners, while Darmerica LLC faced severe penalties for distributing adulterated APIs from unverified suppliers.
Most distributors don't fail because they're running rogue operations. They fail because of documented, fixable gaps — incomplete temperature mapping, missing records, unqualified suppliers, and inadequate personnel training — that appear repeatedly in MHRA deficiency data and FDA enforcement actions.
What follows is a practical breakdown of GDP compliance requirements, the most common audit failure points, and a step-by-step preparation approach — so your next inspection ends with a certificate, not a warning letter.
Key Takeaways
- GDP compliance is mandatory for wholesale distributors under FDA, EU, and WHO frameworks; treating it as optional puts your license at risk
- Audits assess five core pillars: quality management, personnel, storage, transportation, and documentation
- Most audit failures trace back to preventable gaps — temperature mapping, cold chain breaks, missing records, or unqualified suppliers
- Audit readiness means maintaining SOPs, CAPA logs, and self-inspections year-round, not scrambling before an audit
- Your distributor's GDP status directly impacts your own regulatory standing
What Is GDP and Why Does It Matter for Pharmaceutical Distributors?
Good Distribution Practice (GDP) is the set of mandatory standards governing how medicinal products and their active ingredients are sourced, stored, transported, and handled between manufacturer and end-user. Regulatory bodies including the FDA, European Commission, and WHO issue and enforce these standards.
GDP compliance is a legal requirement for wholesale distribution authorization holders. In the European Union, it's mandatory under Article 80(g) of Directive 2001/83/EC. In the United States, wholesale distributors must comply with 21 CFR Part 205 and the Drug Supply Chain Security Act (DSCSA).
GDP vs. GMP: Understanding the Distinction
While Good Manufacturing Practice (GMP) governs how drugs are made, GDP governs how they are moved and distributed. Both fall under the broader GxP umbrella, but compliance obligations for GDP rest specifically with wholesale distributors, not manufacturers.
Certain FDA regulations do overlap. 21 CFR Part 211 establishes cGMP for pharmaceutical manufacturing, yet two of its provisions — § 211.142 (Warehousing procedures) and § 211.150 (Distribution procedures) — are widely referenced as foundational benchmarks for storage and stock rotation. Their legal scope applies to manufacturers, but distributors routinely adopt them as baseline practice.
The Stakes for Non-Compliance
Regulatory actions for GDP failures are severe and immediate:
- FDA Warning Letters and EU import/export bans
- Product seizures and costly recalls
- Wholesale license suspension or revocation
- Loss of client contracts and long-term reputational harm
WHO estimates that more than one in ten medicines in low- and middle-income countries are substandard or falsified. Distributors failing to qualify suppliers or properly manage returns create vulnerabilities that allow counterfeit products into the legal supply chain, triggering regulatory license suspensions.
The Core Pillars of GDP Compliance
GDP compliance — as defined by EU Commission Guidelines (2013/C 343/01) and WHO standards — is organized around five interconnected operational pillars that auditors systematically assess. The US FDA enforces equivalent standards through drug wholesale distributor regulations, with state-level requirements adding further specificity.
Quality Management System (QMS)
A documented QMS is the foundation of GDP compliance. It must include:
- Quality policy with authorized procurement and release procedures
- Quality risk management framework
- Periodic QMS reviews based on risk assessment findings
- Change control procedures covering personnel, facilities, and suppliers
The QMS must demonstrate that compliance is embedded in daily operations, not triggered only by inspections. MHRA deficiency data shows that Chapter 1 (Quality Management) generates the highest frequency of major deficiencies during audits.

A strong QMS also sets the standard for how personnel are trained and held accountable — the next area auditors examine closely.
Personnel Training and Responsible Person Designation
EU GDP Chapter 2 mandates a designated Responsible Person (RP) who oversees all GDP compliance activities. This person must:
- Be continuously contactable and fulfill responsibilities personally
- Possess defined authority to make GDP compliance decisions
- Oversee role-specific training programs
In the United States, the equivalent is the state-level Designated Representative (required in states like California and Florida), who must be physically present during normal business hours and actively involved in daily operations.
All distribution personnel must receive initial and ongoing GDP training covering product security, falsified medicine detection, and SOP adherence. Auditors will request training records and assess whether training is role-specific and routinely evaluated for effectiveness.
Watch for this gap: High staff turnover in warehouse and logistics roles routinely leaves employees handling temperature-sensitive products without adequate GDP awareness — one of the most common personnel-related deficiencies cited in audits.
Storage and Temperature Control
Storage facilities must meet strict requirements:
- Clean, dry, vermin-free warehouses with documented temperature and humidity monitoring
- Segregated storage areas for returned, recalled, suspected counterfeit, and hazardous products
- FEFO (First Expired First Out) stock rotation with documented exceptions
- Regular physical stock reconciliation
Temperature mapping of storage areas must be validated seasonally and revalidated after facility changes. WHO Technical Report Series No. 992, Annex 5, Supplement 8 mandates mapping when areas are both empty and in normal loaded condition, identifying zones where products should not be stored (e.g., near cooling coils).
Where seasonal variations affect storage areas, facilities must conduct mapping twice: once during the warmest season and once during the coldest season.
Transportation and Cold Chain Integrity
Wholesalers must maintain product quality, integrity, and security throughout transit. This includes:
- Validated packaging materials (gel packs, dry ice protocols)
- Real-time temperature logging with calibrated equipment
- Emergency SOPs for transit delays
- Pre-dispatch confirmation of valid delivery orders
- Proper container labeling with handling and storage instructions
The scale of cold chain failure is significant: a systematic review found that 16.7% of transport shipments in developed countries and 35.3% in developing countries experienced freezing temperatures. In studies tracking temperatures across multiple cold chain segments, 75–100% of shipments encountered freezing exposure — most often caused by unconditioned ice packs.
Cold chain breaks during last-mile delivery or transshipment are among the most frequently cited audit deficiencies.

Documentation and Traceability
GDP requires complete, end-to-end traceability of all medicinal products. All records must be:
- Dated and authorized at the time of entry
- Retained for a minimum of seven years (EU) or three years (US)
- Compliant with data integrity standards — no retroactive edits, tamper-evident systems required
Electronic records must comply with FDA 21 CFR Part 11 (for US operations) and EU Annex 11 (for EU operations).
Record types include:
- Dispatch records and receipt confirmations
- Complaints logs and deviation reports
- Temperature and humidity logs
- Supplier qualification files
- CAPA (Corrective and Preventive Action) entries
The Most Common GDP Audit Failures — and How to Prevent Them
Most GDP audit failures cluster around the same handful of issues — temperature mapping gaps, weak supplier qualification, documentation problems, training lapses, and CAPA mismanagement. These aren't obscure compliance edge cases. They're predictable, and that makes them preventable.
Incomplete Temperature Mapping
Many distributors map storage facilities only under peak summer or winter conditions, missing transitional season risks and failing to identify hotspots near loading docks or high-traffic doors.
To address this:
- Implement continuous environmental monitoring with alert thresholds
- Schedule revalidation every two to three years or after any facility modification
- Map both empty and loaded conditions
- Identify and document zones unsuitable for product storage
Inadequate Supplier and Customer Qualification
Relying solely on paperwork without on-site audits or periodic performance reviews leaves critical gaps in supply chain integrity. The FDA Warning Letter to Darmerica LLC cited reliance on conflicting vendor surveys without verifying compliance, resulting in distribution of adulterated APIs.
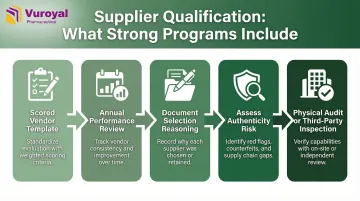
Strong qualification programs share these features:
- Maintain a scored vendor qualification template
- Conduct annual performance reviews
- Document reasoning for all supplier selection decisions
- Assess supplier reputation, product authenticity risk, and pricing patterns that may indicate falsified product
- Require physical audits or verified third-party regulatory inspection histories
Missing, Incomplete, or Non-Compliant Records
Manual documentation systems introduce errors, allow unauthorized changes, and create traceability gaps that auditors spot immediately. Inspection findings consistently cite gaps in contemporaneous temperature records, pencil-written logs, and — in the most serious cases — falsified readings entered retroactively to fill missing data.
The corrective path is straightforward:
- Transition to validated electronic systems that produce audit trails
- Ensure systems meet 21 CFR Part 11 (US) or EU Annex 11 (EU) data integrity requirements
- Implement automated backups and secure storage
- Prohibit manual corrections without documented authorization
Untrained or Inadequately Trained Personnel
High staff turnover — especially in warehouse and logistics roles — results in employees handling temperature-sensitive or controlled products without sufficient GDP awareness.
A defensible training program requires:
- Role-specific onboarding with documented competency assessments
- Refresher training at least annually (quarterly for high-turnover roles)
- Training records maintained for all personnel
- Effectiveness evaluated through practical assessments, not just sign-off sheets
Neglected CAPA Log Management
Auditors routinely inspect the CAPA log to assess how seriously a company takes deviation management. Incomplete entries, unresolved corrective actions, or absence of preventive measures signal systemic compliance weakness — and that signal is hard to walk back during an inspection.
Keep the CAPA log inspection-ready by:
- Reviewing on a defined schedule (monthly or quarterly)
- Ensuring every incident has a documented root cause and closure record
- Demonstrating proactive preventive action planning
- Never closing deviation records before CAPA implementation is confirmed
A Practical GDP Audit Preparation Checklist
Audit readiness requires year-round attention. This checklist gives distributors a structured review framework for both routine self-assessment and the 60-90 days before a scheduled or anticipated inspection.
Pre-Audit Readiness Review
Conduct these self-assessment actions:
- Verify all SOPs are current, approved, and version-controlled
- Confirm the Responsible Person role is formally documented with clear authority to make quality decisions
- Review training records for completeness and recency — check specifically for gaps in personnel files
- Pull the last 12 months of temperature and humidity logs and investigate any gaps or unaddressed excursions
- Conduct a formal mock audit using the PIC/S Aide-Memoire (PI 044-1) or WHO/EU GDP guidelines

Engage an external reviewer for the mock audit — someone unfamiliar with your daily operations will flag procedural gaps your internal team has stopped noticing.
Documentation and Records Audit
Complete this documentation sweep:
- Confirm all dispatch and receipt records are complete and retrievable
- Verify supplier qualification files are current with on-site audit reports
- Check that all CAPA entries are closed or have documented open-item justifications
- Confirm complaints and returns log is properly categorized (product quality vs. distribution complaints)
- Verify evidence of investigation and resolution for each complaint entry
Storage and Equipment Verification
Perform these physical and systems checks:
- Verify temperature mapping validation records are current
- Confirm all monitoring equipment has up-to-date calibration certificates (annual minimum)
- Inspect segregated storage areas (returned goods, recalled product, suspected counterfeit) for proper labeling and access controls
- Conduct stock reconciliation check comparing recorded vs. physical inventory
- Document investigation of any discrepancies
Transportation and Third-Party Logistics Review
Third-party logistics providers (3PLs) are an extension of the distributor's GDP responsibility and must be formally qualified. Pull 3PL qualification files and verify current status across these areas:
- Review cold chain validation records for all shipping containers
- Check deviation reports from the previous 12 months
- Confirm vehicle temperature loggers are calibrated
- Verify emergency protocols for transit delays are documented and communicated to drivers
Self-Inspection and Continuous Improvement Loop
EU GDP Chapter 8 requires a formal self-inspection program. Distributors must select scope areas, assess compliance, document findings, and follow up on corrective actions.
The "snapshot audit" approach:
- Isolate one process area per review cycle
- Simulate a theoretical non-compliance incident
- Test whether the existing process can identify, contain, and document it
- Document findings, assign ownership, and close improvements within a defined timeframe
An active self-inspection history is itself evidence of compliance culture. Auditors respond positively to documented, proactive improvement — it demonstrates that quality management is ongoing, not staged for inspection.
Partnering with a GDP-Compliant Pharmaceutical Distributor
For research organizations, clinical trial sponsors, hospitals, and pharmaceutical manufacturers, the compliance burden does not sit solely with internal operations. The GDP status of the distributor you source from is a direct input to your own regulatory standing.
A distributor that lacks proper licensure, documented quality systems, or cold chain validation can introduce GDP gaps into an otherwise compliant supply chain.
Key Qualifications to Verify
When evaluating pharmaceutical distributors, verify:
- FDA registration and state wholesale distributor licenses
- Drug Business Quality Management Standard Certification
- Cold chain transport capability with validated packaging and continuous temperature monitoring
- Regulatory guidance support for imported drug approval and filing
- Documented QMS with active CAPA and self-inspection programs
- Third-party compliance verification through external audit partnerships
Example: VuRoyal Pharmaceutical
VuRoyal Pharmaceutical checks each of these boxes. The Boston-based company is FDA-registered, holds a Massachusetts wholesale distributor license, and carries Drug Business Quality Management Standard Certification — credentials that map directly to the qualifications above.
In practice, this means cold chain pharmaceutical transportation with validated packaging, free regulatory guidance on imported drug registration and filing, and documented compliance systems built for research organizations, clinical trial sponsors, hospitals, and pharmaceutical manufacturers. For any organization preparing for a GDP audit, a distributor relationship like this functions as a compliance asset, not a liability.
Frequently Asked Questions
What are Good Distribution Practices (GDP) in the pharmaceutical industry and are they a regulatory requirement?
GDP are legally required standards governing the storage, handling, and transportation of pharmaceutical products throughout the supply chain. Compliance is mandatory for wholesale distribution license holders under FDA, EU, and WHO frameworks — not a voluntary best practice.
Is GDP part of GxP?
Yes, GDP is a subset of the broader GxP (Good Practice) umbrella alongside GMP (Good Manufacturing Practice), GCP (Good Clinical Practice), and GLP (Good Laboratory Practice). GDP focuses specifically on distribution operations, while GMP governs manufacturing processes.
What is 21 CFR 210 and 21 CFR 211?
21 CFR Parts 210 and 211 are the FDA's current Good Manufacturing Practice (cGMP) regulations for pharmaceutical manufacturing. While primarily GMP standards, certain Part 211 requirements — including warehousing procedures and traceability records — overlap directly with GDP expectations for storage and receipt.
What is a GMP checklist?
A GMP checklist is a documented audit tool used to verify manufacturing compliance with cGMP regulations. It differs from a GDP checklist, which focuses on distribution-specific requirements such as temperature mapping, cold chain validation, supplier qualification, and traceability records.
What are the consequences of failing a GDP audit?
Failing a GDP audit can trigger:
- FDA Warning Letters or EU import/export bans
- Product recalls and wholesale license suspension or revocation
- Long-term reputational damage with customers and regulators
Recent enforcement actions confirm regulators are actively revoking licenses for data integrity failures and unqualified supplier relationships.
How often should pharmaceutical distributors conduct self-inspections for GDP compliance?
GDP guidelines require a formal self-inspection program on a defined schedule — typically annually at minimum. Many compliant distributors conduct focused "snapshot" audits quarterly on rotating process areas to maintain continuous audit readiness and catch compliance gaps before an inspector does.


