
Introduction
Pharmaceutical manufacturers, generic drug developers, and research organizations share a persistent sourcing challenge: finding verified reference standards that meet exact specifications for dissolution assays and impurity testing. A single substandard material can compromise an entire study, invalidate method validations, or derail regulatory submissions worth millions of dollars.
FDA, EMA, USP, and ICH all require certified, traceable reference standards with complete documentation throughout drug development and batch release testing. Provider selection isn't a procurement formality — it's a regulatory risk decision.
This article covers what verified reference standards require in dissolution and impurity testing contexts, why provider credentials matter as much as product availability, how to evaluate suppliers against regulatory benchmarks, and which red flags to watch for during procurement.
Key Takeaways
- Verified reference standards are traceable, fully characterized materials used as benchmarks in dissolution assays and impurity testing
- FDA, USP, ICH, and EMA require certified standards with assigned purity values and full analytical characterization
- Qualifying providers requires FDA registration, quality management systems, and a complete Certificate of Analysis for each standard
- Using unverified standards risks method validation failures, inaccurate impurity reporting, and rejected regulatory submissions
What Are Verified Reference Standards for Dissolution and Impurity Testing?
Defining Verified Reference Standards
Verified reference standards are highly purified, fully characterized chemical compounds that serve as benchmarks to confirm the identity, potency, purity, or impurity profile of a drug substance or product. According to ICH Q7 Section 11.17, a primary reference standard is a substance demonstrated by extensive analytical testing to be authentic material of high purity, obtained from an officially recognized source or prepared by independent synthesis.
The term "verified" carries specific regulatory weight. Characterization must use multiple independent analytical techniques, typically:
- Nuclear Magnetic Resonance (NMR) for structural confirmation
- High-Performance Liquid Chromatography (HPLC) for purity assay
- Mass Spectrometry (MS) for molecular identity
- Karl Fischer titration for water content
The standard must also arrive with complete traceable documentation: a Certificate of Analysis (CoA), assigned purity value, defined storage conditions, and a traceable lot number.
Role in Dissolution Assays
When a tablet or capsule undergoes dissolution testing per USP <711> and <724>, the resulting solution is assayed using HPLC or UV spectrophotometry. A reference standard for the active pharmaceutical ingredient (API) is required to quantify the drug released. Without a verified API standard, the quantitative dissolution result has no reliable benchmark.
Dissolution results are reported as a percentage of labeled content released over time — the Q-value. The accuracy of the reference standard's assigned purity is therefore directly linked to the reliability of every dissolution result the lab produces.
Role in Impurity Testing
Impurity reference standards detect, identify, and quantify trace contaminants or degradation products in a drug. ICH Q3A(R2) and Q3B(R2) mandate their use in submissions and batch release testing, establishing strict thresholds:
| Maximum Daily Dose | Reporting Threshold | Identification Threshold | Qualification Threshold |
|---|---|---|---|
| ≤2 g/day | 0.05% | 0.10% or 1.0 mg/day | 0.15% or 1.0 mg/day |
| >2 g/day | 0.03% | 0.05% | 0.05% |
Impurities detected at or above these thresholds must be identified and qualified using specific, well-characterized impurity reference standards.
Standard Classifications
Four standard types serve different roles across method validation, routine testing, and regulatory submissions:
- Primary reference standards — highest purity, directly traceable to pharmacopoeial sources (USP, EP, JP); required for method validation and regulatory submissions
- Secondary reference standards — calibrated against primaries; used for routine lab work; ICH Q7 Section 11.19 requires qualification against a primary before first use and periodic requalification
- Working standards — in-house preparations validated against certified standards; manage testing costs while preserving traceability
- Impurity-specific standards — synthesized or isolated impurities used for profiling and qualification at ICH-mandated thresholds

Why "Verified" Matters
A verified standard includes complete documentation that regulators expect during audits:
- Certificate of Analysis with assigned purity value and uncertainty
- Structural confirmation data (NMR/MS spectra)
- Identity confirmation method and purity assay method with results
- Storage conditions and expiry or retest date
- Lot number and authorizing analyst or QA signatory
Without this documentation package, a standard cannot be defended during an FDA audit or regulatory submission — regardless of the chemical's actual purity.
Why Provider Verification Matters in Dissolution and Impurity Testing
Regulatory Stakes
FDA, EMA, and USP require that reference standards used in validated analytical methods be traceable, characterized, and documented. Using an unverified standard—even one that appears chemically correct—can invalidate an analytical method validation or trigger regulatory findings during inspections.
Real-world consequence: In a 2022 FDA Warning Letter to Aspire Pharmaceuticals (MARCS-CMS 630328), the agency cited the firm for using FTIR identity testing by comparing sample spectra to an "unverified reference sample" rather than a qualified reference standard. The FDA explicitly stated: "Identity testing using FTIR to compare the sample spectrum to an unverified reference sample is not an appropriate, scientifically valid method to verify that specifications are met." The firm had improperly used previous vendor lots without performing any qualification work.
Downstream Consequences
Sourcing from unqualified providers leads to:
- Failed dissolution method validation
- Inaccurate impurity profiles
- Incorrect reporting of impurity levels against ICH thresholds
- Product recalls
- Rejection of ANDAs or NDAs
- Extensive Out of Specification (OOS) investigations requiring retesting
Traceability is Non-Negotiable
Every verified reference standard must have a traceable audit trail from manufacture through characterization, certification, and storage. ICH Q7 Section 11.17 mandates that the source of each primary reference standard be documented, and manufacturers must maintain records of each standard's storage and use per supplier recommendations.
Purity Assignment Accuracy
Impurity reference standards require an assigned purity value derived via quantitative NMR (qNMR) or mass balance determination, not a nominal estimate. This distinction matters because purity assignment directly drives every downstream calculation.
An unverified standard with an incorrect purity assignment causes systematic errors across every assay that uses it — potentially leading to incorrect conclusions about whether impurity levels exceed regulatory thresholds.
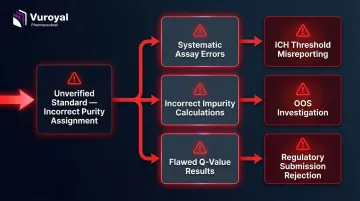
What GMP/GLP Compliance Requires from Suppliers
Providers who supply reference standards to regulated laboratories must operate under appropriate quality management systems. USP <11> and ICH Q7 set clear expectations for qualified suppliers:
- Demonstrated regulatory credentials (FDA registration, ISO accreditation, or equivalent)
- Documented quality management systems with audit-ready records
- Complete analytical characterization data accompanying each lot
- Defined storage and handling protocols aligned with standard stability requirements
Types of Reference Standards: Matching the Right Standard to Your Test
Dissolution Assay Requirements
Dissolution assays express results as a percentage of labeled content (Q-value), so they require quantitative API standards—either primary or secondary—with strictly assigned purity values derived via qNMR or mass balance.
Before each run, system suitability standards confirm that instrument calibration and method performance meet acceptance criteria. Without this verification step, Q-value results carry no regulatory weight.
Impurity Testing Requirements
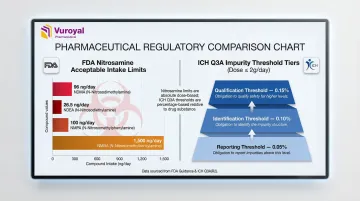
For ICH Q3A/Q3B-compliant profiling, impurity reference standards must support detection at three regulatory thresholds for doses ≤2 g/day:
- Reporting: 0.05%
- Identification: 0.10%
- Qualification: 0.15%
Nitrosamine impurities sit in a separate, more demanding category. Under FDA's September 2024 guidance, these compounds carry ultra-low Acceptable Intake (AI) limits that require purpose-built standards:
| Nitrosamine | FDA Acceptable Intake Limit |
|---|---|
| NDMA | 96 ng/day |
| NDEA | 26.5 ng/day |
| NMPA | 100 ng/day |
| NMBA | 1,500 ng/day |
Validating LC-HRMS methods at these trace concentrations demands highly pure reference standards. Isotopically labeled variants are typically required for accurate quantitation.
Qualitative vs. Quantitative Standards
The distinction between these two standard types determines what a result is actually worth in a regulatory context:
| Standard Type | Assigned Value | Typical Use |
|---|---|---|
| Quantitative | Yes — with uncertainty | Dissolution assays, impurity limit calculations, batch release |
| Qualitative | No | Structural confirmation in early development only |
Qualitative standards are appropriate during candidate screening and method development. Once a program moves toward regulatory submission or commercial batch release, quantitative standards with documented assigned values are required.
Critical Criteria for Evaluating Reference Standards Providers
Regulatory Registration and Credentials
Providers should be FDA-registered entities (for US supply) or hold equivalent regulatory standing in their jurisdiction. FDA registration as a pharmaceutical wholesaler demonstrates accountability and audit readiness.
Example: VuRoyal Pharmaceutical, as an FDA-registered, Massachusetts-approved pharmaceutical wholesaler with drug business quality management standard certification, exemplifies the regulatory credentialing that organizations should expect from a trusted supplier.
Certificate of Analysis Completeness
Every shipment must include a CoA meeting ICH Q7 requirements (Sections 11.41-11.44):
Required CoA elements:
- Material identity, grade, batch number, and release date
- Each test performed with acceptance limits and numerical results
- Date and signature of authorized quality unit personnel
- Original manufacturer's name, address, and telephone number
- For repackers/distributors: reference to original manufacturer and copy of original batch certificate
Red flag: CoAs that obscure the original manufacturer's identity or lack complete analytical test results are non-compliant with ICH Q7 and should be rejected.
Analytical Characterization Depth
Qualified providers characterize standards using multiple orthogonal techniques:
- NMR to confirm molecular structure
- HPLC or GC to determine purity
- MS to verify molecular identity
- Karl Fischer titration for water content (where relevant)
Reject providers supplying standards with only a single analytical data point. FDA guidance requires standards to be "thoroughly characterized to assure identity, strength, and quality" — meaning one technique is never sufficient for a qualified reference standard.
Supply Chain Integrity and Cold-Chain Logistics
Temperature excursions during shipping or storage can degrade standards, altering their assigned purity. USP <11> mandates that standards be stored in original stoppered containers away from heat and protected from light. NIST SP 260-136 warns that organic analytes can change rapidly due to degradation.
Critical for:
- Nitrosamine standards (highly susceptible to photolytic decomposition)
- Compounds susceptible to hydrolysis
- Certain API standards requiring refrigeration
Providers must demonstrate:
- Validated cold-chain logistics capabilities
- Temperature-controlled storage (typically 2–8°C or below −20°C)
- Temperature excursion documentation on delivery
- Quarantine protocols for shipments lacking temperature logs

Custom and Hard-to-Source Impurity Standards
Cold-chain capability addresses storage and transit — but sourcing gaps are a separate challenge. Not all impurity standards are commercially available through pharmacopeias, so providers with custom synthesis partnerships or access to specialized chemical suppliers can source or develop standards for:
- Unique impurities in novel APIs
- Newly identified degradants
- Nitrosamine Drug Substance-Related Impurities (NDSRIs)
For innovator and generic drug programs, this capability covers the full impurity lifecycle: from initial identification through certified standard supply.
Navigating Global Sourcing and Regulatory Compliance
International Procurement Challenges
Importing reference standards internationally involves:
- Complex import/export regulations
- Customs documentation requirements
- Risk of counterfeit or degraded materials through unverified channels
- Potential shipment seizures for documentation failures
FDA Import Alert 66-40 targets foreign establishments with significant GMP violations. Products from firms on the "Red List" are subject to Detention Without Physical Examination (DWPE), meaning shipments are detained and refused admission unless the importer proves the specific product is not violative.
Benefits of Certified Importers
Organizations benefit from working with certified importers who can:
- Manage customs declarations and electronic entry filings via Automated Commercial Environment (ACE)
- Ensure cold-chain continuity across borders
- Provide regulatory guidance for jurisdiction-specific filing requirements
- Maintain complete traceability documentation per ICH Q7 Section 17.2
Example: VuRoyal holds certified importer status and provides free regulatory guidance alongside active logistics management. The company handles imported drug approval registration, import filing, customs clearance, and cold-chain transportation — covering the full procurement cycle from origin to receiving lab.
Documentation Requirements for International Shipments
Certified importers manage this documentation burden directly, but labs and procurement teams should understand what's required for each shipment:
- Import permits specific to the receiving country
- Country-of-origin certifications (Certificate of Origin)
- Customs declarations with FDA Establishment Identifier (FEI)
- Manufacturer name, address, and facility details
- Authentic Certificates of Analysis (CoAs) from the original manufacturer
- Affirmations of Compliance for expedited FDA review

Failure to maintain proper import documentation results in shipment seizures, supply delays, and testing program interruptions. In a Warning Letter to Creative Essences, Inc. (MARCS-CMS 710658), FDA cited the firm for relying on suppliers' CoAs without establishing supplier reliability, noting the lack of scientific evidence that components conformed to specifications.
Frequently Asked Questions
Frequently Asked Questions
What qualifications should a verified reference standards provider have for dissolution testing?
Providers must hold FDA registration or equivalent regulatory standing, quality management certification (such as drug business quality management standard certification), and the ability to supply complete CoA documentation with traceable purity assignments derived from validated analytical methods.
What is the difference between a certified reference standard and a working standard in dissolution assays?
Certified reference standards carry full analytical characterization and pharmacopoeial traceability required for method validation and regulatory submissions. Working standards are in-house preparations validated against certified standards for routine daily testing — lower cost, but with fully documented traceability.
How do USP and ICH guidelines govern the use of reference standards in impurity testing?
USP Chapter <11> and ICH Q3A/Q3B set purity thresholds — 0.05% reporting, 0.10% identification, 0.15% qualification for doses ≤2 g/day — along with documentation requirements and the need for qualified impurity standards at or above each threshold. Any standard used must be evaluated and characterized for its specific intended application with an assigned purity value.
What documentation should accompany verified reference standards for regulatory submissions?
Required documentation includes:
- CoA with assigned purity value and measurement uncertainty
- Structural confirmation data (NMR/MS spectra) and identity confirmation method
- Stability or retest data, lot traceability records, and storage condition validation
- Purity assay method with results and authorized QA signatory approval
Can the same impurity reference standard be used for both dissolution assay and impurity profiling?
The application determines the required standard type. Dissolution assays need quantitative API standards with assigned purity for Q-value calculations, whereas impurity profiling requires dedicated impurity standards with separate purity assignments — the same compound cannot serve both purposes without verified characterization data supporting each use.
How should verified reference standards be stored to maintain their integrity for dissolution and impurity testing?
Follow the provider's specified storage conditions: typically controlled temperature (2–8°C or below −20°C depending on the compound), with protection from light and moisture. USP <11> mandates storage in original stoppered containers away from heat and humidity. Never return unused portions to the original container.